南开大学本科生破译武汉2019冠状病毒基因组

能直接看论文的,可以到这里下载
还有一个中国知网的链接
请大家支持中文期刊《生物信息学》
以下大家看文章,由于某些人的举报与恶意攻击,我不做任何解答,已做出的解答不得已删除了
武汉2019冠状病毒基因组的生物信息学分析
陈嘉源1,施劲松2,丘栋安3,刘畅4,李鑫1,赵强1,阮吉寿5,高山1*
1南开大学生命科学学院, 天津300071;
2东部战区总医院, 南京 210016;
3英国诺丁汉特伦特大学生物科学系, 诺丁汉 NG11 8NS;
4南开大学医学院, 天津300071;
5南开大学数学科学学院, 天津300071
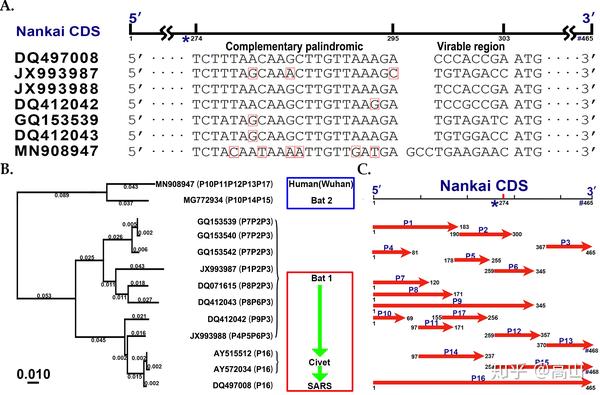

图1. 武汉2019冠状病毒的起源与可变翻译
Fig1. Origin and alternative translation of the Wuhan 2019 human coronavirus genome
A:Nankai CDS是beta冠状病毒基因组中一段高度保守的序列,包括一段22 bp的互补回文序列(用*表示)。Nankai CDS还包括一个8或11 bp的可变区,分别属于两种不同长度(是465或468 bp,用#表示)的Nankai CDS;B:进化树构建使用13条去除可变区的Nankai CDS。使用多种方法进行进化分析的结果一致,这里只展示邻接(Neighbor Joining,NJ)法的结果:武汉2019冠状病毒源自中华菊头蝠某个群体(蓝色方框内),但与SARS冠状病毒差异巨大(红色方框内);用P1到P17(见图1C)可以对13个beta冠状病毒进行基因分型(小括号内),其结果与进化分析的结果一致。C:可变翻译导致从Nankai CDS可以预测出至少17个蛋白质,分别命名为P1到P17。这里展示的是使用一个起始密码子ATG和三个终止密码子进行蛋白质预测的结果,使用四个起始密码子(ATG、GTG、CTG和TTG)和三个终止密码子会预测更多的蛋白质,但不改变可变翻译相关结论。
A: The 22-bp complemented palindrome (named Nankai complemented palindrome) and the CDS (named Nankai CDS) have a high degree of evolutionary conservation in betacoronavirus genomes. A 8- or 11-bp long variable region in Nankai CDS resides in two types of Nankai CDS. The first type has a length of 465 bp, while the second type has a length of 468 bp. B: Consistent results were obtained by different phylogenetic analysis methods. Here, we only show the result using the NJ (Neighbor Joining) method. The Wuhan 2019 human coronavirus (in blue box) with large differences from the SARS coronavirus (in red box), may originate from betacoronaviruses in Chinese horseshoe bats. Phylogenetic analysis and genotyping (in parentheses) of 13 viruses using 17 putative proteins (Fig 1C) achieved consistent results. C: The putative proteins (named as P1 to P17) were predicted from the Nankai CDS using ATG as the start codon, and TAA, TAG and TGA as stop codons. The prediction using ATG, GTG, CTG and TTG as start codons, and TAA, TAG and TGA as stop codons did not change all conclusions in the present study.