
如何快速估算测序量?

考虑这样一个问题,如果要保证基因组上95%的区域覆盖深度在30x以上的话,那么最低的测序深度应该是多少?
其实这类关于测序量估计的问题,对于做生物信息的人来讲应算是家常便饭了。多数时候我们都能直接根据以往项目的经验来获得,或是说的更具体些,在单个人的变异检测中一般要有25x以上的覆盖度才能得到一个比较靠谱的结果,于是以此为目的地给出测序量的估计值。
少数缺乏有效数据的情况下也会有直接拍脑袋拍出一个值来的疯狂行为,但虽说是拍脑袋,其实也不是随便拍的,拍脑袋的背景靠的是身后丰富的经验。相对更好一些的估计方式就是直接模拟数据,不过总是用模拟数据还是让人觉得麻烦,最好是能不用花多少时间,也不用做很多的计算就能脱口给出的。因此,我想在这里说一下这种情况下我的解法。当然并不一定会完全准确,仅作交流,欢迎各位拍砖。
闲话说完,回到上面的问题,在不通过数据模拟也不拍脑袋的情况下,要如何才能估算出一个合理的值呢?其实在作出任何推断之前我们都应当要先有一个合理的前提假设,或者说是理论依据来作为后续分析的基础。
我们都知道短序列测序的一个特点是:理论情况下,位点被覆盖到的深度符合泊松分布(测序没什么问题的话,实际的情形也相差不多),但实际上在这种情况下用正态分布来考虑也是合理的,作为一个估计值,误差也是能够接受的,这是我们的基础。之所以想用正态分布来考虑,是因为正态分布有许多方便于计算的性质。其中一个很有用的法则,就是68-95-99法则,意思就是距离均值一个标准差的区域围起来的面积大约是总体的68%,2个标准差的区域范围的面积是总体的95%,3个标准差区域范围占到了总体的99%,如果你自己想要验证这一法则也并不困难,只需做些积分就能算出来。如下图,均值用μ表示,标准差用σ表示。
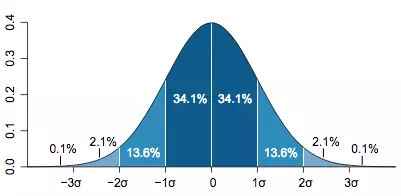
现在事情就很简单了,从图中我们可以看出,只要30x深度的位置在−2σ以下,那么就能达到理论的要求。要得到这一结果,问题就只剩下一个了,此时我们只需要知道测序深度分布的标准差就能粗略估计出此时我们所需要的最低平均测序深度。虽然这个标准差跟许多因素有关,我们这里用illumina公司的HiSeq测序平台为例子,依照以往基因组重测序的经验,σ约等于10x。那么,简单算一下,此刻,理论上我们需要测50x就可以使得基因组上有97.7%的区域其覆盖深度都在30x以上了,注意这里不是95%了,因为我们的区域实际上是[−2σ,+∞),而不是[−2σ,+2σ]! 再除掉一些边边角角的误差,50x这个值在这里应当是合理的了。
以上计算都是以正态分布为基础而做出的估计。当然了,如果一定要用泊松分布去推算也可以,只是运算起来会麻烦很多。此外,如果是不同系列或是不同公司的测序仪,σ就不一定是10了。
如果喜欢更多的生物信息和组学文章,欢迎搜索并关注我的微信公众号“碱基矿工”(ID: helixminer)
