
基于结构的药物设计:从分子对接到分子动力学

背景
近年来,计算机辅助药物设计(Computer-Aided Drug Design,CADD)方法在药物研发中不断崭露头角,如已批准上市的卡托普利、沙奎那韦、利托那韦和茚地那韦等,其早期研究大大受益于CADD方法。CADD已成为药物研发流程中不可或缺的一部分。

基于结构的CADD计算方法通过解决两个主要任务来支持苗头化合物的发现和结构优化:1、预测小分子与蛋白质靶标结合;2、计算结合亲和力。本文首先回顾了分子对接,然后概述了高通量分子对接虚拟筛选。最后,讨论了分子动力学(Molecular Dynamics,MD)模拟。基于MD模拟方法的使用正在稳步增加,因为它们最适合分析配体结合的热力学与动力学。
分子对接
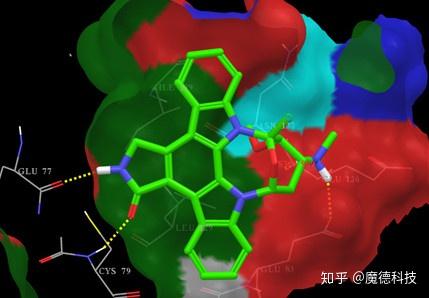
分子对接主要用来确定蛋白质靶标中小分子的最佳位置和取向。尽管该方法的成功依赖于靶标和软件,但也与不良的结合亲和力相关。蛋白质-配体相互作用的质量可以在一定程度上由配体效率(Ligand efficiency, LE)表示,即每个配体的非氢原子的平均结合能。然而,大多数分子对接预测的研究偏向于结合蛋白质靶标的分子具有可检测的亲和力和可用的晶体结构。基于大约300种激酶抑制剂的研究表明,简单的评分函数(仅范德华力)在拟合结合亲和力值方面优于总能量(即范德华力和静电力),但用于高通量分子对接进行计算筛选的预测能力较差。计算模拟的真正挑战是以足够的准确度计算相对结合能,使得用于体外测试的化合物的最终选择中存在尽可能多的真阳性;反过来,结合能的成功评估依赖于对结合模式的准确预测。
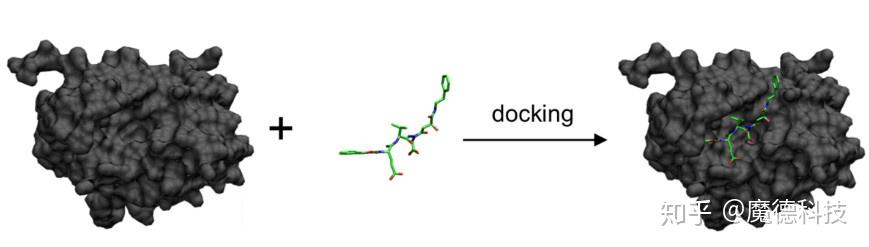

基于分子对接的虚拟筛选
虚拟筛选的原理是评估分子库中化合物是否有可能与蛋白质结合,并列出最有可能以最高亲和力结合的分子。虚拟筛选的主要挑战不是识别小分子库中的少数纳摩尔抑制剂,而是减少通过体外分析选择用于验证的化合物子集中的假阴性化合物数量。很少有研究能够系统地分析分子对接的成功率,即正确预测结合蛋白质靶标的化合物的百分比。虽然许多论文中展示了很不错的命中率,但是定义的命中标准总是主观的依赖于研究。虚拟筛选成功案例必须谨慎对待,因为选择过程通常涉及具有重要专业知识的研究人员的目视检查。为了正确地对不同软件的性能进行基准测试,应该引入通用标准,并且应该最小化人为干预,因为结合模式分析的复杂性或与体外验证的成本相关。


分子对接中的主要挑战
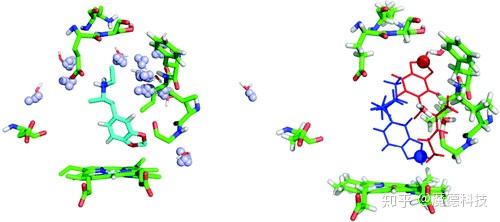
溶剂处理:保守的水分子
水分子的处理仍然是一个挑战,因为很难预测哪些溶剂分子在结合位点是必须的,哪些可以被进入的配体取代。结合位点水分子的稳定性可以基于多晶体结构的分析来确定,或者使用明确的溶剂进行MD模拟确定。

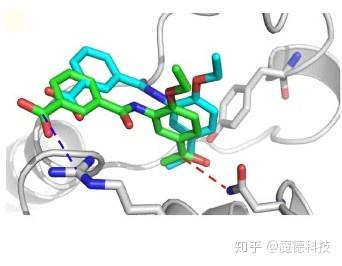
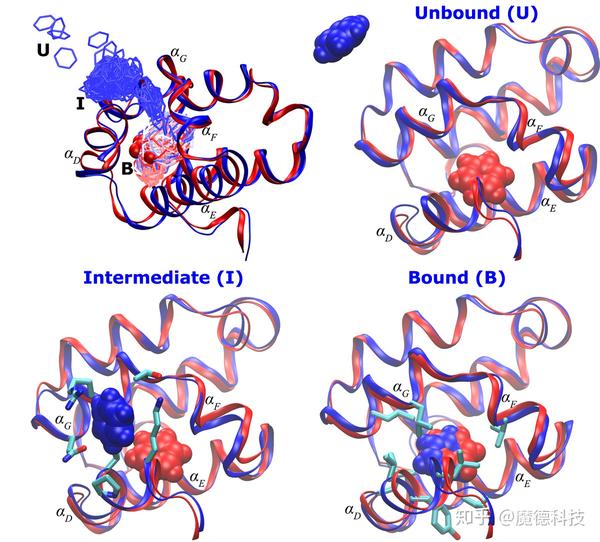
结合位点的柔性
蛋白质的柔性是计算机辅助药物发现的另一个挑战。自从结构生物学方法的出现及其在基于结构的药物发现中的应用以来,许多蛋白质结合位点不能表示为单个帧,因为蛋白结构经常发生显著的重排以适应不同的配体。蛋白质结构上的结合位点的柔性信息可从实验或模拟研究中获得。
分子动力学模拟和受体柔性
长期以来,研究人员都提出MD模拟能够提供超越蛋白质结晶学可用的蛋白质构象,并发现新的隐蔽结合位点,扩大靶标的成药性。
分子动力学诱导拟合利用全原子模拟来增加或减少活性位点口袋的大小。诱导结合的配体的初始姿势能够通过将已知抑制剂对接到靶蛋白的口袋获得。可以在较低浓度配体存在的情况下进行MD模拟,其已被证实能够有效地探测构象空间和发现新的隐蔽口袋,随后进行实验验证。
配体结合和解除结合的MD模拟
MD模拟在药物发现和设计中的应用不断扩大。MD模拟可以用于映射配体结合位点和分析结合模式。配体解除结合的模拟提供了对复合物亲和力的深入理解,并量化了整个过程中的能量变化。据研究报道,未结合的速率依赖于蛋白质受体的状态,而蛋白质受体的状态又取决于实验条件,特别是配体的浓度。因此,由于小配体的解除结合可能比蛋白质的构象变化快一个数量级,故在执行高通量分子对接时选择正确的构象状态至关重要。配体结合/解除结合动力学也可能在药理学中具有重要意义,因为复合物的滞留时间被提出比药物亲和力本身更准确地预测药物的功效。

参考资料
1. Śledź P, Caflisch A. Protein structure-based drug design: from docking to molecular dynamics. Current Opinion in Structural Biology, 2017, 48:93.
